

Abstract
Neuroendocrine tumours of the pancreas can secrete numerous peptides, leading to various recognizable clinical syndromes. The secretion of pancreatic polypeptide has been used as a marker for neuroendocrine tumours but is considered to be a biologically inert peptide. A 37-year-old woman had watery diarrhea syndrome from pancreatic polypeptide hyperplasia. Only 2 other reported cases in the literature have described pancreatic polypeptide hyperplasia; however, this is the first reported case in which the patient was successfully treated by surgical resection, with a 2-year follow-up. This report and review of the literature illustrate that pancreatic polypeptide hypersecretion may present as a clinical endocrinopathy.
Ectopic hormonal secretion by islet cell tumours has produced distinctive clinical syndromes. Many of these syndromes have helped us define the pathophysiology of the peptides themselves. It was during the purification of insulin that pancreatic polypeptide (PP) was discovered.1 A clinical syndrome resulting from hypersecretion of PP has not been readily discernible because a biologic role for PP has not been found. Many case reports have demonstrated that PP-secreting tumours can cause a watery diarrhea (WDHA) syndrome (Verner–Morrison syndrome) similar to vipomas.2–6 There has been only 1 reported case of WDHA syndrome caused by PP hyperplasia.7 Reported here is a case of PP hyperplasia successfully treated with surgical bulking, lending support to the thesis that PP is not always a silent peptide.
Case report
A 37-year-old woman presented with sudden onset of watery diarrhea. She was having between 10 and 20 bowel movements daily. Initially, her condition was thought to be infectious, so she was given metronidazole (500 mg every 8 hours orally) for 12 days. Because her diarrhea persisted, she was admitted to hospital 2 weeks later with dehydration and weight loss. She was given fluids intravenously and allowed nothing orally; however, the watery diarrhea persisted. Her family and medical histories were noncontributory. There was no history of laxative abuse or previous gastrointestinal abnormalities. There was no personal or family history suggestive of multiple endocrine neoplasia. Findings on colonoscopy were normal, as were stool cultures, and the serum levels of vasoactive intestinal polypeptide (VIP), 5-hydroxyindoleacetic acid, gastrin and calcitonin. Her initial electrolyte levels were were in keeping with hypokalemic metabolic acidosis which was corrected with intravenous fluids. An abdominal CT scan demonstrated a left ovarian cyst. One month later, ultrasonography showed that it had resolved. To rule out an intestinal lymphoma or an occult carcinoid, an upper gastrointestinal series with follow-through was done, and CT scans of her chest and head were obtained. All findings were normal. Findings on endoscopic ultrasonography of the pancreas were also normal.
She was started on somatostatin analogue (octreotide), 100 mg 3 times a day. Her bowel movements decreased within 2 days to 3 formed stools daily, and further decreased to 1 daily formed bowel motion within a week. When the octreotide was with-drawn the watery diarrhea returned within 24 hours and only resolved once the octreotide was restarted.
A diagnosis of secretory diarrhea secondary to an islet cell tumour not yet identified was made and exploration of the pancreas undertaken. The pancreas was completely mobilized, but no tumour was found on bimanual palpation. Intraoperative ultrasonography also did not reveal any tumours within the pancreas. A biopsy specimen of a small piece of the tail of the pancreas was obtained, which on frozen-section examination was suggestive of islet cell hyperplasia. A three-quarter distal pancreatectomy was performed. Postoperatively, the patient did well. Her initial serum fasting PP levels postoperatively were 75 pg/mL (range from 1 to 100 pg/mL). She was discharged 7 days later on no medication and was having 1 to 2 formed bowel movements daily.
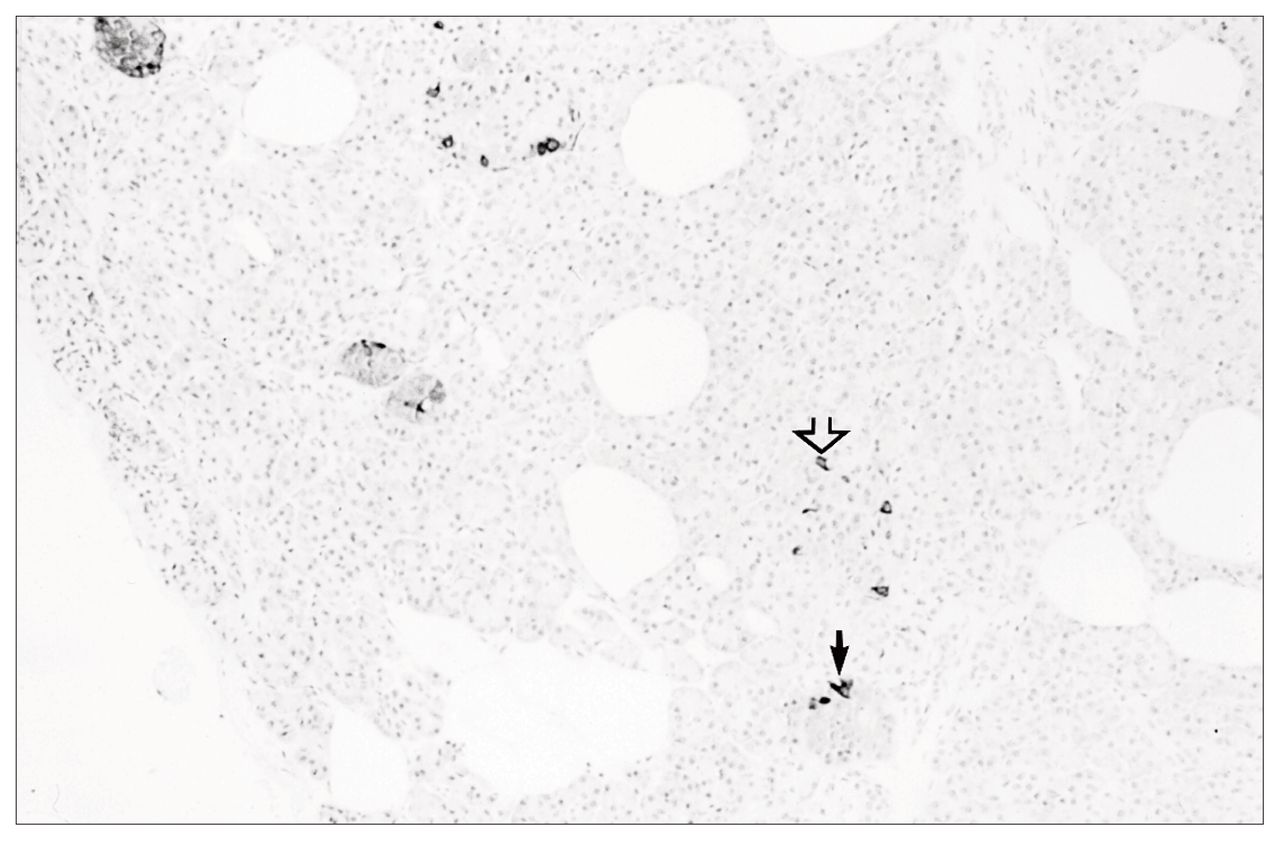
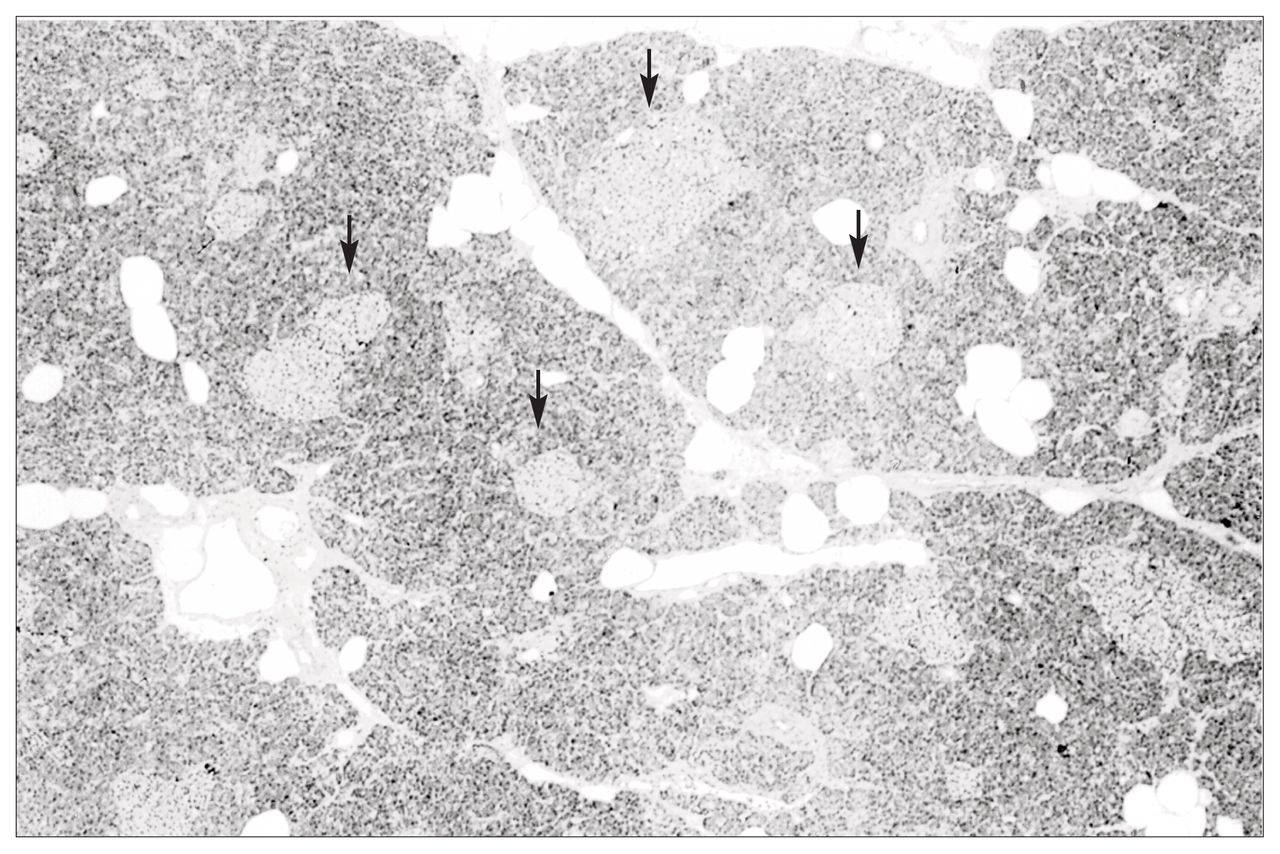
Pathological examination of the excised specimen revealed pancreatic islet cell hyperplasia (Fig. 1). The pancreas demonstrated an increase in both the size and the number of islets compared with normal pancreas. Immunohistochemical staining demonstrated no excessive staining of insulin, glucagon or somatostatin-secreting cells within these abnormal islets. The pancreas stained negative for gastrin and VIP. However, cells staining for PP were readily identified within islets and isolated in small nests throughout the pancreas (Fig. 2). This distribution of PP-staining cells was interpreted as being abnormal. In the normal pancreas PP cells are predominantly found in the head of the pancreas and are not seen in such abundance in the body and tail as in this patient. This histologic appearance was consistent with pancreatic islet cell hyperplasia, staining predominantly for PP.

Pancreatic islet cell hyperplasia. Note the increased number and size of the pancreatic islets (arrows) (hematoxylin–eosin stain, original magnification × 40).
{kind=link}
{kind=link}
Immunohistochemical staining positive to pancreatic polypeptide both within the islets (small arrow) and along the ducts (open arrow) (pancreatic polypeptide stain, original magnification × 40).
At follow-up, 36 months postoperatively, the patient was still well. She had 1 to 3 formed bowel movements daily. She was taking only pancreatic enzymes, with each meal. The fasting serum glucose level remained normal. Her serum fasting PP level remained normal for 3 years, but the most recent PP level was slightly elevated at 272 pg/mL (normal range from 64 to 243 pg/mL).
Discussion
Very little is known about the pathophysiological role of PP in humans. Fasting levels increase with age, prolonged fasting, uncontrolled diabetes and exercise.6 Ingestion of proteins, fat and carbohydrates are usually followed by a rise in PP that is sustained for 4 to 8 hours. This response to meals can be abolished by atropine.8 Greenberg and associates8 suggested that PP inhibits gallbladder contraction and pancreatic enzyme secretion. For this reason, many believe that PP islet cell tumours are clinically silent; with the addition of this case, there are now 22 cases reported in the literature. 6 The mean age of the patients was 51 years (ranging from 20 to 74 years). Diarrhea was recorded in 7 of these patients, and 5 patients had steatorrhea. The other 10 patients presented with silent tumours found incidentally. Eight (36%) of the patients presented with metastases to the liver or lymph nodes, or both. PP hyperplasia, including this case, was found in only 3 of these reported cases.
The majority of tumours are solitary, and some have been associated with the multiple endocrine neoplasia, type I, syndrome. Further complicating the picture is the fact that PP cell hyperplasia has been frequently reported in association with other functioning islet cell tumours.9–11 PP cells are increased in 20% to 67 % of functioning and nonfunctioning tumours of the pancreas.6 It is for this reason that serum PP levels have been utilized as a screening tool in patients with suspected islet cell tumours.11 There is, of course, the possibility that a small islet cell tumour was missed on sectioning of the specimen and the PP hyperplasia was just a reaction to this islet cell tumour. However, both endoscopic and intraoperative ultrasonography as well as bimanual palpation of the pancreas at laparotomy failed to demonstrate any abnormalities. Therefore, this case appears to demonstrate that PP cell hyperplasia in isolation from other endocrine-secreting tumours can cause a WDHA syndrome. Tomita and colleagues7 also reported 2 cases of PP hyperplasia, 1 causing a WDHA syndrome and 1 clinically silent.
This case not only illustrates the difficulty with present day technology in localizing islet cell tumours, it also illustrates the difficulties in diagnosing islet cell hyperplasia intraoperatively. The PP cells of the normal pancreas are more abundant in the head region and are distributed both within and outside the islets.12 On frozen section, the islets seen in this case were larger in size and in number than normal pancreas, suggesting hyperplasia. Grossly, the pancreas was normal, but immunohistochemical staining demonstrated an overabundance of PP cells throughout the pancreas and within the islets themselves. The response to somatostatin preoperatively confirmed a secretory form of diarrhea. Debulking the pancreas allowed for a 3-year diarrhea-free period. Intraoperatively, it is difficult to define the extent of the surgical resection when faced with islet cell hyperplasia. Removal of too much pancreas may lead to pancreatic insufficiency with respect to both exocrine and endocrine functions. Dividing the pancreas at the level of the superior mesenteric vein usually allows for normal endocrine and exocrine function if the remaining pancreas is normal. Although this patient had pancreatic insufficiency postoperatively, her endocrine function remained intact. The rise in the PP level 3 years later is not surprising, since we believe that the remaining pancreas also had PP hyperplasia and with time there will be a return of WDHA syndrome.
It is unfortunate that preoperative serum levels of PP were not obtained in this patient. However, the clinical response to octreotide, then to surgery, along with the pathological findings, confirm the secretory nature of the diarrhea.
Half of the neuroendocrine tumours arising from the pancreas have been thought to be nonfunctioning. However, the nonsecretory nature of these tumours may be related more to inability to detect secretion or secretion of a biologically inert peptide. PP was once thought to be a biologically inert peptide; however, growing literature supports the fact that this peptide can cause a WDHA syndrome and is not always clinically silent. Treatment of PP-secreting tumours causing endocrinopathies should be similar to other functioning islet cell tumours. Despite nonlocalizing preoperative studies, surgical exploration and intraoperative ultrasonography done by an experienced endocrine surgeon is recommended for all functioning neuroendocrine tumours. Surgical debulking or removal of an isolated tumour may result in excellent palliation, if not cure, of the clinical endocrinopathy.
Footnotes
Abstract presented at the Canadian Association of Gastroentrologists, Banff, Alta., 1995.
- Accepted October 3, 1997.