


Abstract
Objective: To identify outcomes and prognostic variables that predict survival outcomes in adult Wilms tumour patients.
Methods: We collected data on 128 patients with adult Wilms tumour treated between 1973 and 2006. Six cases from our 2 Canadian centres have not been previously reported. We collected data on the remaining 122 patients from published case reports or case series. Analyzed factors included age, sex, favourable or unfavourable histopathology, clinical stage (I, II, III or IV) and chemotherapy and radiotherapy received. The outcomes studied included overall survival (OS) and disease-specific survival (DSS). Univariate analysis with Kaplan–Meier actuarial methodology and multivariate analyses with Cox regression were used to determine outcomes and predictive clinical factors.
Results: The patients’ mean age was 26 (range 15–73) years. After a mean follow-up of 54 (range 2–240) months, the OS and DSS of the entire cohort were both 68%. Favourable histopathology predicted superior OS and DSS (both p < 0.001). Higher clinical stage predicted inferior OS and DSS (both p < 0.001).
Conclusion: Adult Wilms tumour has a poorer prognosis than pediatric Wilms tumour. In adults with Wilms tumour, more aggressive patient- and tumour-specific surveillance and adjunctive therapies than those advocated by pediatric National Wilms Tumor Study guidelines may be warranted, especially in patients with an unfavourable histopathology and higher clinical stage.
Wilms tumour is the most common primary renal malignancy of childhood. Cure rates in the pediatric population of patients with clinically organ-confined or advanced stages of disease are high when a multimodal approach involving surgery, chemotherapy and radiotherapy is used.1 The extent and timing of surgery has been based on studies by the National Wilms Tumor Study Group (NWTS), and the extent of disease and histology of the primary tumour has directed the adjuvant therapies.1
Adult Wilms tumour is rare, and its clinical presentation in patients can often be indistinguishable from renal cell carcinoma or other more common adult renal tumours.2–6 There has been controversy over the prognosis in cases of adult Wilms tumour.7–10 Owing to its low incidence, 2–5 the prognostic variables have not been studied in a large cohort of patients. In our experience, adults with Wilms tumour had poor overall survival despite treatment according with contemporary pediatric NWTS protocols.11 The survival outcomes for adult Wilms tumour have never been shown in a large cohort of patients to be clearly inferior to outcomes for pediatric Wilms tumour. We hypothesized that adults with Wilms tumour have poorer outcomes, and our objective was to identify outcomes and prognostic variables that predict survival outcomes, using the collected data on all published reports of adult Wilms tumour patients.
Methods
Data were collected on 128 patients with adult Wilms tumour treated between 1973 and 2006.12–20 Patients in these series were treated according to protocols based on contemporaneous NWTS recommendation. Six cases from 2 Canadian centres (Queen’s University and the University of Western Ontario) have not been previously reported. We obtained data for the remaining 122 patients from published case reports or case series.12–20 We collected information about age, sex, favourable (FH) or unfavourable (UH) histopathology, clinical stage (I, II, III or IV), chemotherapy and radiotherapy.
On the basis of the NWTS results, Wilms tumour is divided into FH or UH depending on the absence or presence of anaplasia.21 Anaplasia has been defined by the NWTS as the combination of cells with very large hyperchromatic nuclei and multipolar mitotic figures. The enlarged nuclei must be at least 3 times as large as the typical blastemal nuclei in both axes, and hyperdiploid mitotic figures must be present.22 The NWTS has established a staging system for Wilms tumour (Box 1).22 We used these definitions to determine clinical stage and histopathology type.
National Wilms Tumour Study Group staging system
| Stage | Extent of tumour |
|---|---|
| I | Tumour is confined to the kidney and completely resected. Specific criteria
|
| II | Tumour extends locally outside the kidney, but is completely resected. Specific criteria
|
| III | There is residual tumour confined to the abdomen without hematogenous spread. Specific criteria
|
| IV | There are hematogenous metastases. |
| V | Tumours are present in both kidneys. |
In addition, we investigated data on overall survival (OS) and disease-specific survival (DSS). We used Kaplan–Meier actuarial methodology to estimate OS and DSS. The logrank test was used to compare the significance of differences between the survival curves. Cox regression analysis was used to determine which factors were an independent predictor of survival outcomes.23 We considered a value of p < 0.05 to be statistically significant.
Results
Patients who presented with Wilms tumour had a mean age of 26 (range 15–73) years. After a mean follow-up of 54 (range 2–240) months, the OS and DSS of the entire cohort were both 68%. Of the patients, 117 received multiagent chemotherapy, 3 received single-agent chemotherapy, and 7 received no adjuvant chemotherapy. Information about chemotherapy was not recorded for 1 patient. The most frequent chemotherapeutic regimens used were vincristine and actinomycin-D (43 patients); vincristine, actinomycin-D and adriamycin (48 patients); etoposide, carboplatinum, ifosfamide and adriamycin (11 patients); and vincristine, actinomycin-D and epirubicin (6 patients). For 77 patients, radiotherapy targeted the renal fossa (1000–5000 cGy), lungs (1200–2000 cGy) and/or local recurrences (5000 cGy). A further 50 patients did not receive any radiotherapy, and this information was not available for 1 patient.
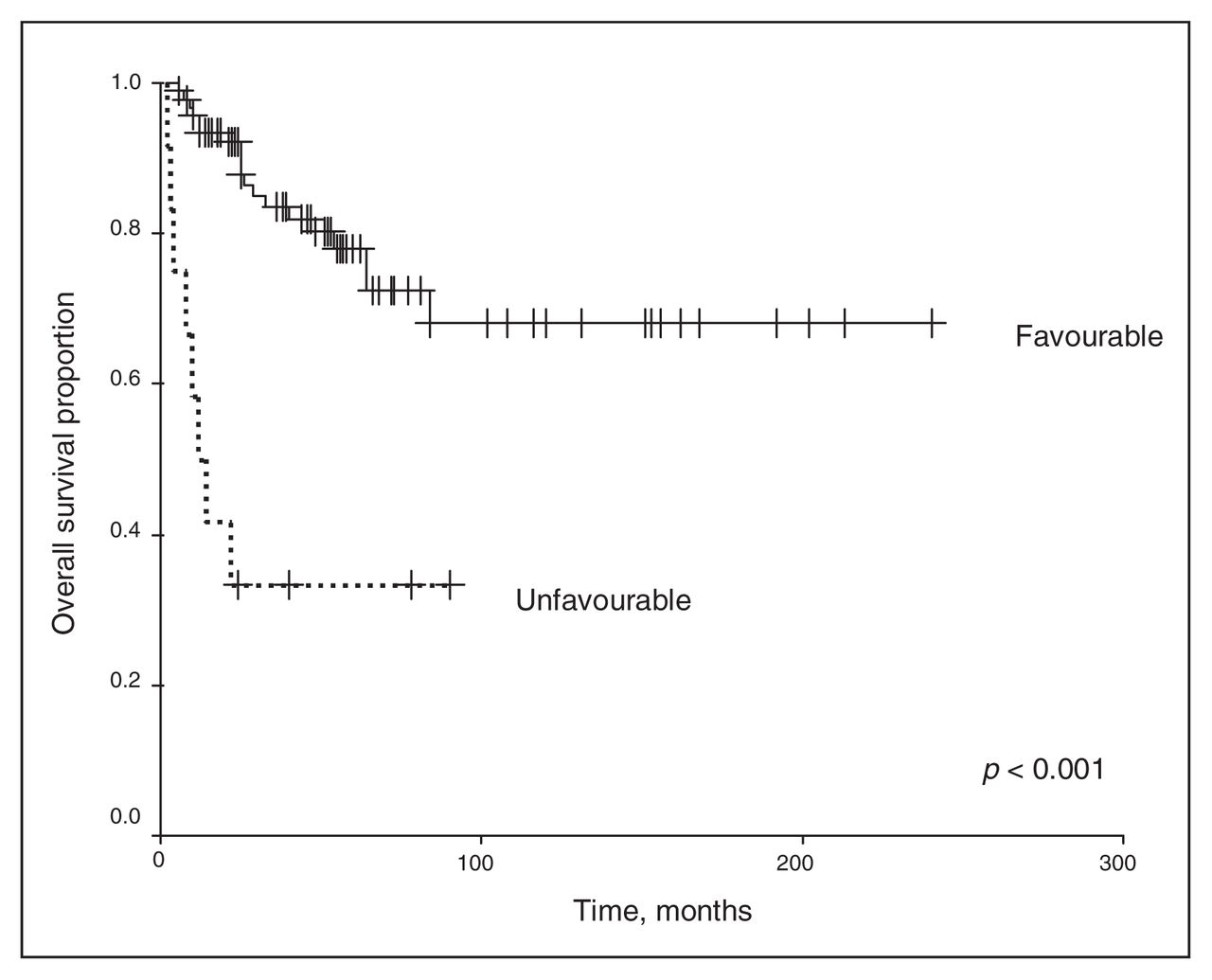
We excluded chemotherapy from the analysis because of the small number of patients receiving any regimen other than the vincristine and actinomycin-D–based therapies. Univariate analysis revealed that the clinical stage and histopathology of the primary tumour were associated with survival outcomes. As shown in Figure 1 and Figure 2, a higher clinical stage was associated with inferior OS and DSS (p < 0.001 for both). As shown in Figure 3 and Figure 4, UH was associated with inferior OS and DSS (p < 0.001 for both). On univariate analysis, adult age, sex and therapy with external beam radiation were not associated with any survival outcomes. On multivariate analysis, histopathology was independently associated with OS and DSS (p = 0.001 and p = 0.003, respectively), and stage was independently associated with OS and DSS (p = 0.013 and p = 0.015, respectively). Table 1 and Table 2 summarize the results of the univariate and multivariate analyses.
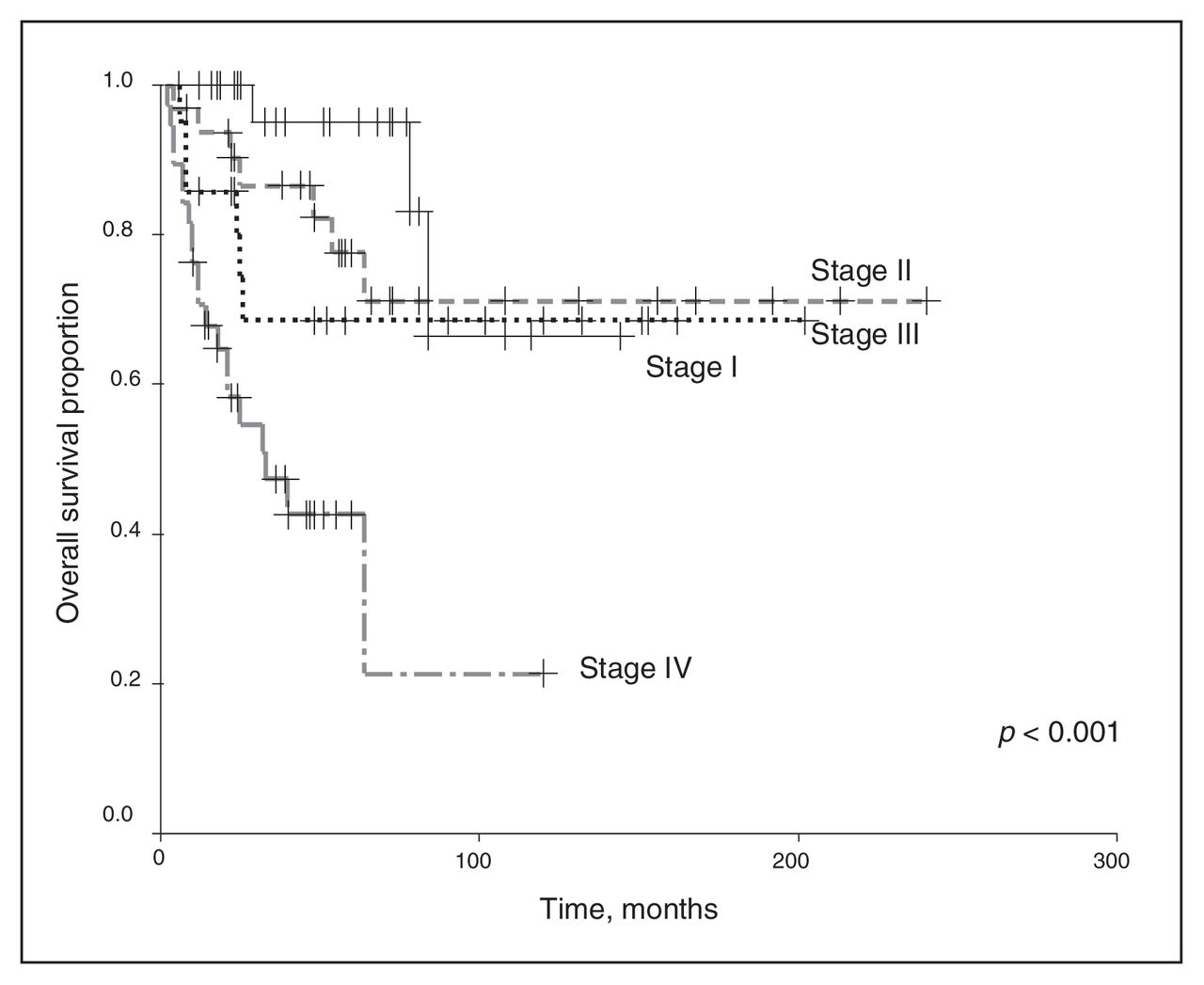
Overall survival for the 128 patients, stratified by disease stage.
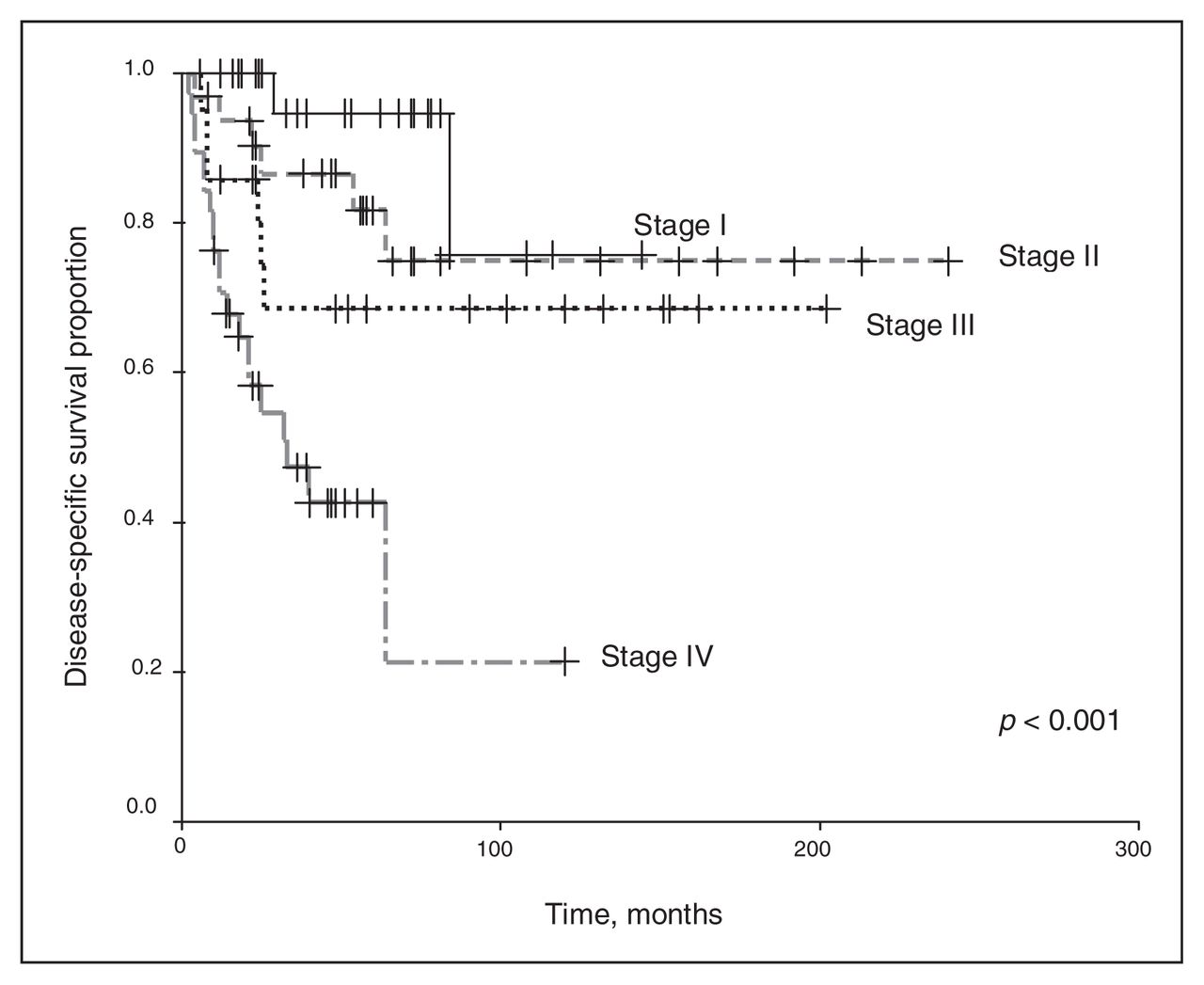
Disease-specific [death specific to Wilms tumour] survival for the 128 patients, stratified by disease stage.

Overall survival for the 128 patients stratified by favourable versus unfavourable histology.

Disease-specific [death secondary to Wilms tumour] survival for the 128 patients stratified by favourable versus unfavourable histology.
Age, sex, histopathology, tumour stage and radiotherapy received as predictors of overall survival in univariate and multivariate Cox regression analyses
Age, sex, histopathology, tumour stage and radiotherapy received as predictors of disease-specific survival in univariate and multivariate Cox regression analyses
Because the numbers were insufficient, we were not able to stratify patients with UH for stage-based comparisons. The overall outcomes in these patients were poor, with a 37% 5-year overall survival.
Discussion
In the past, adults with Wilms tumour have been treated with therapeutic protocols based on data derived from pediatric NWTS data. However, owing to small numbers in each of the adult Wilms tumour case series, it is not clear whether the prognosis of adult Wilms tumour is comparable to that for pediatric Wilms tumour. We believe that our study adds novel data showing the prognosis to be significantly worse in cases of adult Wilms tumour. In pediatric patients studied in NWTS III, D’Angio and colleagues24 showed that the risk of relapse from FH Wilms tumour is low, at about 10% for stage I–II tumours and 20% for stage III–IV tumours. Conversely, all UH tumours had inferior prognosis, compared with FH tumours, regardless of stage (36%–45% 5-year survival). In our study, overall patient survival was only 65% in patients with FH and 37% in patients with UH. Because we only reviewed studies that treated patients with adult Wilms tumour according to pediatric NWTS protocols, it is not clear why overall results in adult Wilms tumour appear to be inferior when compared with pediatric Wilms tumour, despite stage-and histology-based comparisons.
It is possible that our results are biased by diagnostic error. Adult Wilms tumour might have been wrongly diagnosed in several of the series that we used for our analysis. Indeed, some of these patients may have had sarcoma and not Wilms tumour. However, there are some important trends that must be highlighted. As in the pediatric NWTS results, for FH tumours, the prognoses for stage I and II tumours were not different, and similarly, the prognoses for stage III and IV tumours were identical. As well, among adult Wilms tumour patients with UH, there was a marked reduction in survival, compared with survival rates seen in the FH group. Again, this is in keeping with prognostic trends seen in the pediatric NWTS results. Nevertheless, we recommend that histology from patients with presumed adult Wilms tumour be sent to the NWTS for confirmation of diagnosis.
In addition to the potential difficulties of differentiating adult Wilms tumour from sarcoma, the diagnosis of anaplasia and staging can be difficult. 22 The tumour must be sampled well as small foci of anaplasia can be associated with a poor response to therapy.21 Extensive gross sectioning and good histologic preparations, fixations, sectioning and staining are absolutely required to recognize anaplasia, and thus UH, correctly.22 Most Wilms tumour with anaplasia occurs in older children and adults, and any anaplasia present, identified or unidentified, results in significantly poorer survival outcomes. Staging challenges arise because the tumours usually distort the renal pelvis, hilar structures and renal capsule. Thus, differentiating between stage I and II tumours can be extremely difficult.22
The poor results for adult Wilms tumour may also be due to the adult oncologist’s lack of familiarity with this disease. In fact, as an offshoot of the NWTS, Kalapurakal and colleagues10 reviewed 23 patients with FH adult Wilms tumour. Their overall 5-year survival rate was excellent (83%) and suggested that therapy in adults with FH should be directed according to pediatric NWTS protocols. 10 The patients analyzed in our study were treated between 1973 and 2006, and treatment protocol varied depending on the NWTS recommendations at the time. Conversely, the adults treated in the Kalapurakal study were treated by contemporary NWTS IV and V protocols, and the excellent results may reflect improvements in diagnostic testing and therapy. Nevertheless, we believe that it is important to include the input of pediatric oncologists in the treatment of adults with Wilms tumour.
Even with a collaborative approach between pediatric and adult oncologists, there will be challenges in adapting pediatric chemotherapy treatment dosages to adults because of the toxicity of these high dosages. However, the use of hemopoietic growth factors to allow dosage escalation and to decrease the toxicity of systemic chemotherapy may be considered for certain patients at high risk for treatment failure with the recommended NWTS IV and V protocols.
The extent of surgical excision including the regional lymph nodes may also be a factor in survival outcomes. Regional lymph node dissection (LND) is not routinely performed in adult patients with Wilms tumour. One of the 6 patients studied from our Canadian centres did have a recurrence in a regional lymph node requiring re-exploration and regional LND. In adult patients with Wilms tumour, many of the tumours are large and locally advanced at the time of diagnosis. Routine regional LND may improve local recurrence rates and survival. Although this is controversial in renal cell carcinoma, Wilms tumour does have different biology, and routine regional LND has provided survival benefits in other malignancies.25 It is also important to note that, in adults, the diagnosis of Wilms tumour is never made before such therapeutic intervention as radical nephrectomy and lymphadenectomy. Thus patients may not benefit from preemptive chemotherapy, and intervention may be performed outside the state-of-the-art recommendations.
An alternative explanation for the different results in our study may be the heterogeneity in the biology of adult Wilms tumour. In children, Wilms tumour usually presents as an asymptomatic abdominal mass. However, in adults, most patients have a constellation of symptoms that can include flank pain, weight loss or a decline in their performance status.19 This may be secondary to a higher rate of clinically occult metastases. Heterogeneity in biological behaviour also exists in Wilms tumours, whether FH or UH or within the same stage of disease. In addition to the histologic and stage-based prognostic factors used to determine therapies, methods to further define the natural history of a tumour or the response to therapy for individual Wilms tumours will be required. Molecular and genetic studies such as aspiration cytology and cytogenetic analyses of Wilms tumours26 will be necessary to gain further insight into the biology of individual tumours and may provide a means by which to improve outcomes with therapy that is more aggressively patient- and tumour-specific.
Footnotes
Presented at the Canadian Urological Association Meeting, June 26 to July 1, 2004, Whistler, BC, and the NES of the American Urological Association Meeting, May 21–6, 2005, San Antonio, Texas.
Competing interests: None declared.
Contributors: Drs. Izawa, Winquist and Luke designed the study. Drs. Izawa, Al-Omar, Winquist, Steele and Siemens acquired the data, which Drs. Izawa, Winquist, Rodrigues, Luke and Mr. Stitt analyzed. Drs. Izawa, Al- Omar, Luke and Mr. Stitt wrote the article, which Drs. Izawa, Winquist, Rodrigues, Steele, Siemens and Luke reviewed. All authors gave final approval for publication.
- Accepted November 7, 2006.
References
In this issue
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.