


Abstract
Aseptic loosening after total joint replacement remains the most common reason for long-term implant failure. Macrophages activated by submicron wear particles of the polyethylene liner used in joint replacement have been shown to be the source of periprosthetic bone loss. Understanding the role of material chemistry in macrophage activation and the subsequent effects that macrophage-derived enzymes play in the degradation of implanted biomaterials is key to developing methods for prolonging the lifespan of implantable materials.
Total joint replacement is the most successful method of treating end-stage arthritis1,2 and significantly improves the quality of life and functional capabilities of patients suffering from this disease.3 Currently, an estimated 500 000 total joint arthroplasties are performed each year in North America alone, and 30% of these are performed in patients younger than 65 years.4,5 Aseptic loosening after joint replacement, due to bone destruction around the prosthesis, has been clearly established as the main cause of implant failure.6–9 The initial success of total joint replacement has led to an expansion of the indications for the procedure, with younger, more active people benefiting from it.10 The overall 10-year success rate for total joint replacement is 90%, leaving 10% of patients who require revision surgery.4 The probability of revision increases inversely with age (7.5% for those aged 65 to 70 years, 13% for those between 59 and 65 years, and an unacceptable 27% of those 59 years or younger at the time of surgery).1 Revision surgery is technically more challenging and poses higher risks for the patient with respect to the surgery and subsequent complications than primary joint replacement. In addition, at a cost of about $70 000 each, revision surgery adds millions of dollars annually to the cost of our health care system (Dr. Allan E. Gross, Department of Surgery, University of Toronto: personal communication, 1998). Therefore, preventing or reducing the need for revision would have significant economic and social benefits.
Mechanical, biologic and chemical factors contribute to aseptic loosening. The most widely accepted theory for this is that chronic inflammation induced by biomaterial wear particles leads to osteoclastic activation and periprosthetic bone loss. Controversy exists over the mechanism of inflammation and the role of macrophages and osteoclasts in bone loss around the implant. In this article we will discuss the local biologic effects of biomaterial wear particles generated by the implant articulation, specifically, the local effect of wear particles on macrophage activation, the potential of biomaterial degradation and how these factors influence periprosthetic bone loss and implant stability.
Local effects of biomaterial wear particles
Macrophage activation
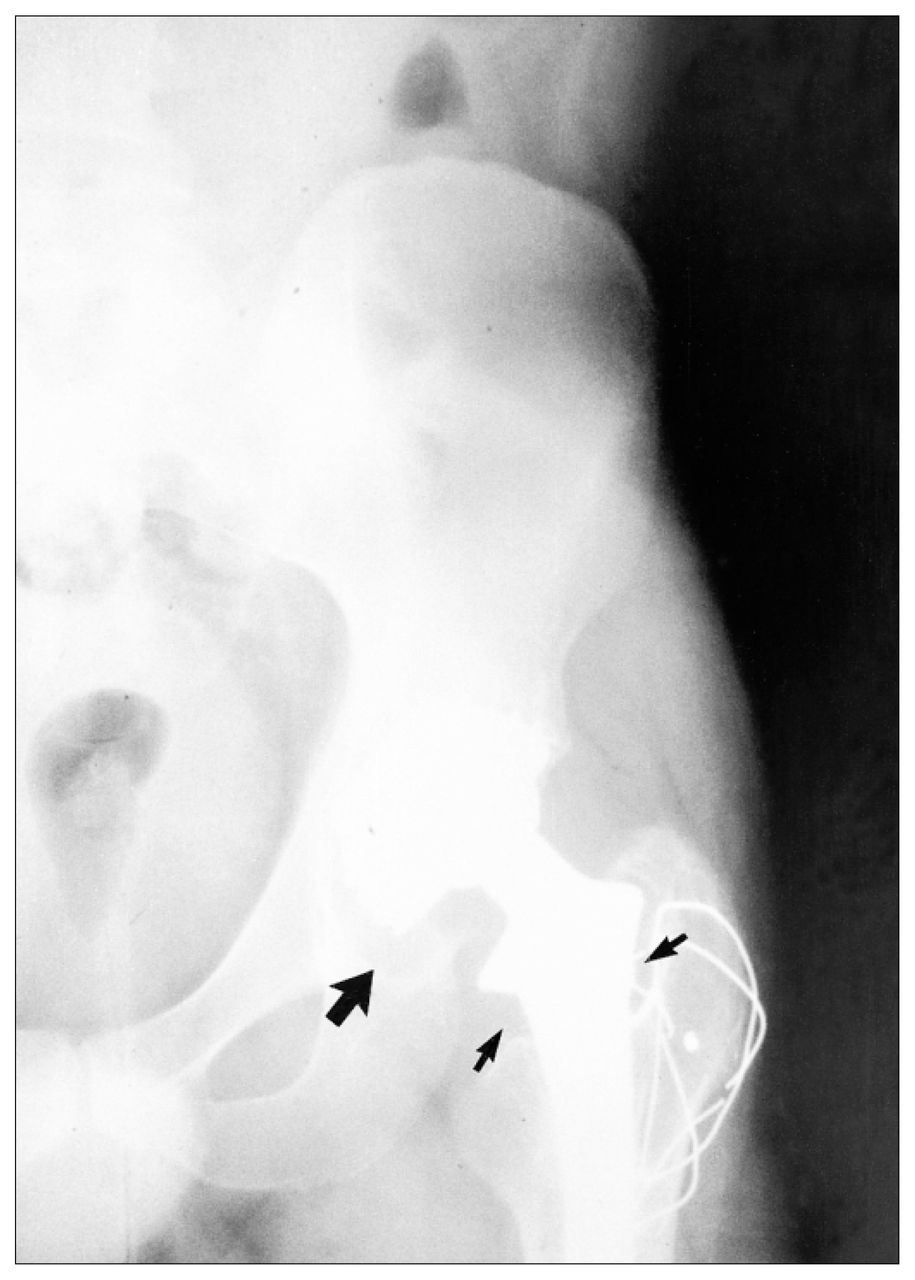
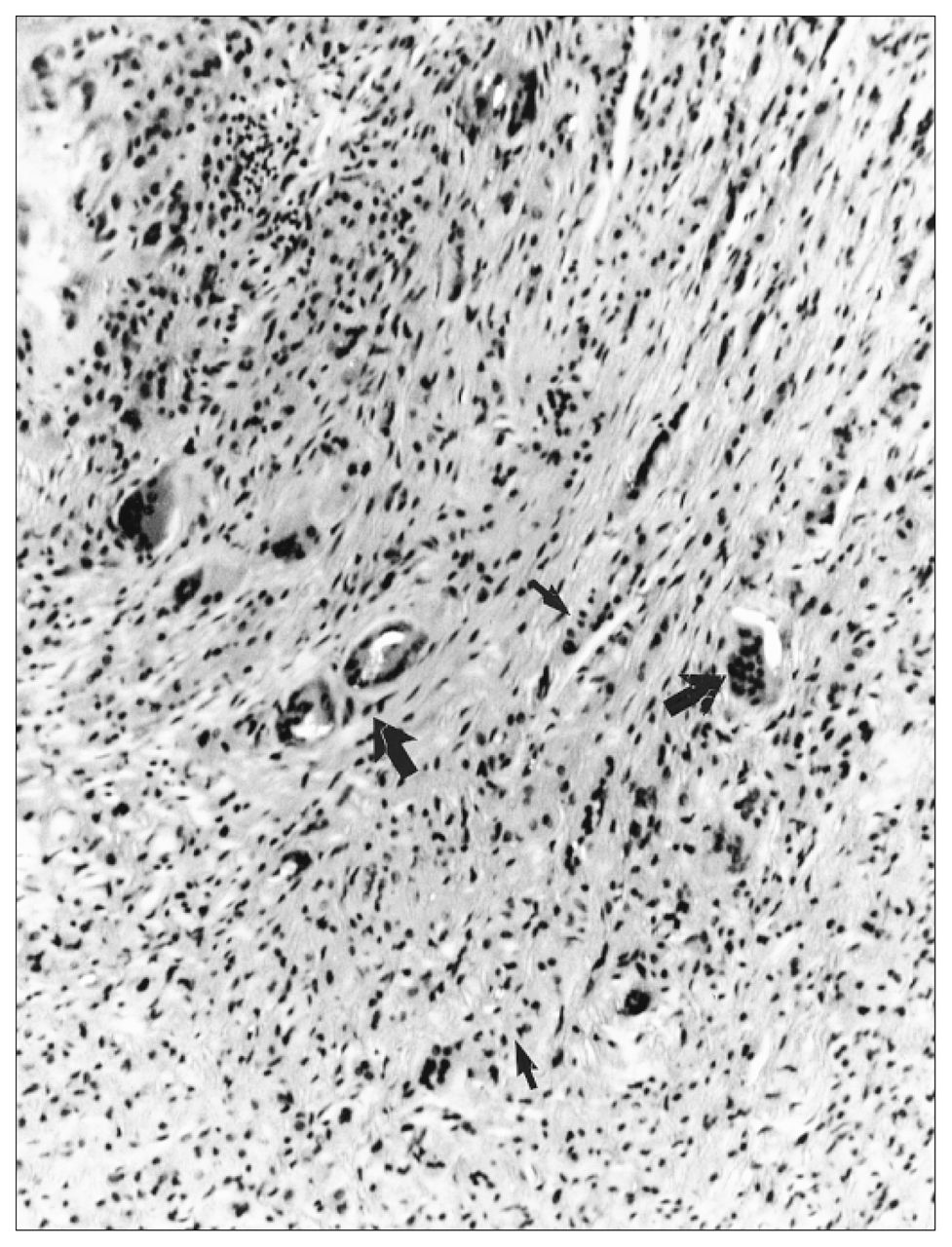
Aseptic loosening or periprosthetic bone loss is diagnosed radiographically as either a linear, diffuse radiolucency or more localized, scalloped lucency, which is referred to as osteolysis (Fig. 1).5,10 At revision surgery, the periprosthetic bone loss observed radiographically contains an inflammatory membrane. Histologic analysis of the inflammatory membrane obtained at revision has clearly identified a chronic inflammatory process, with fibrosis, numerous macrophages, fibroblasts and areas of necrosis associated with particles of wear debris (Fig. 2).6,9,11

Radiograph of total hip arthroplasty demonstrating periprosthetic bone loss around the femur (small arrow) and migration of the acetabulum (large arrow) 3 years after implantation.

Light microscopic examination from a biopsy in the region of periprosthetic osteolysis reveals numerous macrophages (small arrow) and foreign-body giant cells (large arrow) associated with wear particles of polyethylene (bright under polarized light), fibrosis and chronic inflammation (hematoxylin–eosin, orginal magnification × 40).
Immunohistochemical investigations to identify subtypes of cells confirm mature-tissue macrophages as the most predominant cell associated with wear particles. T cells constitute approximately 10% of the total cell population and likely play an important role in modulating the inflammatory response to wear particles by a nonspecific immune mechanism.12 B cells are rare in the inflammatory membrane.13,14
Immunohistochemical and in situ hybridization studies have confirmed the presence of proinflammatory cytokines interleukin 1 (IL-1), tumour necrosis factor alpha (TNF-α), and interleukin 6 (IL-6) in the inflammatory membrane.15,16 In addition to cytokines, prostaglandin E2 (PGE2),17 metalloproteinases and collagenase18 are secreted by cells associated with wear particles. All of these factors could indirectly lead to bone loss by stimulating osteoclastic activity (cytokine), while others (collagenase) directly affect periprosthetic bone loss. These mediators perpetuate inflammation by attracting monocytes to the implant bed, stimulating differentiation to macrophages and engulfment of more wear debris. These cytokines also stimulate osteoclastic differentiation and therefore may influence bone remodelling around the implant.19,20
All wear particles potentially can activate a macrophage. These include metal,21,22 ultra-high-molecular-weight polyethylene (UHMWPE),9,11 polymethylmethacrylate (PMMA),6,23 ceramic24 and hydroxyapatite.25 Digestion of the inflammatory membrane reveals polyethylene (PE) as the most abundant particle, followed by metal and PMMA.26,27 The majority of PE particles are less then 1 μm in dimension; however, particles as large as 200 μm are present. Owing to different mechanical wear mechanisms, debris generated by knees appears generally larger than that generated by hip implants. 28,29 Particle size and volume are important because particles less than 25 μm in dimension will be engulfed, whereas those greater than 25 μm will be surrounded by foreign-body giant cells (FBGCs).19,30 The biologic responses to wear debris of different particle sizes may differ, resulting in adverse affects on local bone remodelling owing to different levels of cytokine secretion and a variable capacity for bone resorption when comparing FBGCs and macrophages.19
The inflammatory membrane retrieved at revision surgery is heterogeneous, with different amounts of particles, different particle types, sizes and patient susceptibility to inflammation. Therefore, a model was developed to introduce PE particles to undifferentiated human monocytes and mature macrophages.31 By glueing PE particles to a glass coverslip with type I collagen we can reliably introduce PE particles to adherent human monocyte-macrophages. When peripheral blood lymphocytes are harvested and “plated” in vitro, the majority are immature monocytes. After 3 to 5 days in vitro the monocytes differentiate to mature macrophages. Alternatively, human monocytes were allowed to differentiate on polystyrene plates over 14 days, then were “trypsinized” and used for particle experiments.32 Studies with the 2 human cell populations were performed: (1) monocytes plated directly onto coverslips or (2) mature trypsinized macrophages were cultured with collagen:PE and control coverslips. After phagocytosis of PE particles, light microscopic examination, scanning electron microscopy and transmission electron microscopy do not show evidence of cytotoxicity, with more than 95% cell viability of either the monocytes or macrophages at 24 hours. Despite initially high cell viability with both cell types, the mature macrophage could be cultured for much longer than undifferentiated monocyte-macrophages (more than 30 days versus 10 days).33 In addition, 2.5 times more mature macrophages cultured with PE survive and form greater numbers of FBGCs when compared with cells cultured with collagen alone. This interesting observation may highlight the relative inertness of simple PE; however, the increased longevity of the macrophages stimulated by PE may play a crucial role in periprosthetic bone resorption, as macrophages have been shown to be capable of direct bone resorption (see below). Initial studies of wear particles with undifferentiated monocytes and mature macrophages confirm cell activation with increased secretion of IL-1 but not IL-6 or TNF-α on PE particle stimulation.33
In summary, macrophage interactions with small particles of simple polyethylene do not appear to be cytotoxic in vitro and indeed lead to prolonged cell survival. The formation of multiple FBGCs and the release of proinflammatory mediators (IL-1), which perpetuate inflammation and accelerate bone loss, were observed over the long-term with cells stimulated by PE. Evidence has emerged that macrophages and FBGCs are capable of direct bone resorption, thus these cells may be responsible for periprosthetic bone loss observed over long periods in vivo.
Chemical degradation of PE
Little is known about the influence of wear-particle chemistry on macrophage activation and thus periprosthetic bone loss. It has been well established that UHMWPE components are oxidized through processing at high temperatures and pressure, 34 sterilization by gamma irradiation,35,36 storage of the irradiated UHMWPE components37 and the implantation environment.38 Oxidation of UHMWPE negatively influences the wear properties of the implant.39,40 Analysis of the PE components after implantation reveals the presence of metal ions.41 Since PE particles are derived from the implant surface, it is likely that the PE particle chemistry has similar changes with oxidation and metal ion association. Polymer–cell interaction studies show that the oxidation of polymeric surfaces has a marked effect on the material’s ability to elicit different cellular responses.42,43 Modification of etched carbon surfaces to increase oxidation results in more intense phagocytosis. Many polymers, including PE, are hydrophobic; several studies have confirmed poor cellular adhesion to hydrophobic surfaces.44,45 Treatment of a hydrophobic surface (polytetrafluoroethylene) with gas plasma resulted in improved wettability and increased cell adhesion.46 These authors also noted increased protein adsorption that likely has a significant effect on cell adhesion mechanisms. In addition, modification of surface characteristics also plays a role in cytokine (IL-1, IL-6, TNF-α) secretion with significant inhibition and decreased adhesion of monocyte-derived macrophages on neutral as opposed to cationic surfaces.45,46 It is apparent that the material surface will influence protein adsorption, which will subsequently influence cell surface receptor interactions and cell adhesion, and will potentially initiate different intracellular cascades affected by the specific signal, cell type and state of cell differentiation. We have developed techniques to chemically modify PE particles so as to simulate changes in wear particle chemistry for use in our monocyte-macrophage culture system.
Not only will the surface chemistry of the material effect cell function, but also long-term exposure of the material to macrophages-derived enzymes can lead to biomaterial degradation. Following the initial interaction and engulfment of the chemically altered PE particle, the biomaterial will become exposed to macrophage-derived lysosomal enzymes with the potential for material degradation. Monocyte-derived macrophages generate reactive oxygen species known to oxidize a variety of biomaterials. Macrophages actively synthesize cholesterol esterase (CE), which has so far been shown to be the most degradative enzyme with many biomaterials as substrates (polyester, polyether and polycarbonate urethanes).47,48 Our in vitro studies show that mature macrophages stimulated by PE particles release significantly more CE than control groups, so chronic exposure to elevated levels of CE could lead to PE degradation. Indeed, preliminary investigations of CE effects on PE have demonstrated significant release of PE degradation products.49 Long-term exposure of PE particles to degradative enzymes could lead to the release of products that could adversely effect bone remodelling around the implant.
These exciting preliminary results have opened a new field of study in joint replacement failure. The identification of macrophage-derived enzyme products of PE will allow alterations in the manufacturing of the material by altering cross-linking, changing processing agents or catalysts, or adding antioxidants to change the profile of the degradation product. Once the degradation products are well characterized, then the toxic effects on specific cell populations, osteoblasts, osteoclasts and macrophages can be determined to analyse how bone loss is created.
Bone remodelling around an implant
Ideally, fixation of total joint replacements may be achieved when either bone is bonded directly to the cement mantle or the implant. Normal bone is formed by osteoblasts, which lay down an unmineralized matrix consisting of type I collagen and proteoglycans that subsequently becomes mineralized. Once the osteoblast is surrounded by mineralized matrix it is referred to as an osteocyte. Osteoclasts, which are derived from macrophages, are responsible for bone resorption. During normal bone remodelling, new bone formation and bone resorption are coupled. When bone is resorbed, factors are released that signal osteoblasts to the area and stimulate new bone formation.20,50 Bone loss may occur due to increased osteoclastic activity, decreased osteoblastic activity or a combination of both.51,52 Macrophages play a role in normal bone remodelling that is altered in a number of pathologic conditions (e.g., tumour metastases), where it is observed that macrophages may directly resorb bone.53 Understanding the cellular mechanisms of periprosthetic bone loss will have important implications directing pharmacologic intervention.
It is clear that wear particles induce inflammation at the bone–implant interface after total joint replacement. The link between macrophage activation and loosening of the prosthesis is bone resorption. What is not clear at present is the role that osteoclasts and macrophages play in the bone loss associated with this inflammation. It is difficult to obtain tissue directly in contact with the bone from revision surgery and therefore, histologic analysis of the retrieval tissue has demonstrated conflicting findings. Earlier studies claimed that the bone loss resulting from wear particles was a result of cytokine activation of osteoclasts. This hypothesis has been well supported by numerous studies in the literature, dating back to the original observation of Goldring and associates6 that the inflammatory membranes secreted high levels of collagenases and PGE2. They demonstrated that conditioned media from interfacial membranes stimulated bone resorption in mice calvaria.17,19 IL-1 has also been shown to be an important stimulator of osteoclastic bone resorption by stimulating osteoclast proliferation, increasing the size of the clear zone and ruffled border, features of an osteoclast that enhance bone resorptive activity. PGE2 plays an important role in regulating bone activity by stimulating osteoclastic bone resorption.54 PGE2 is elevated in response to PE stimulation in vitro.55 Horowitz and Gonzales,19 Haynes and associates21 have suggested that wear debris (PMMA, metal and PE) stimulates macrophages to secrete PGE2 and IL-1, which then stimulate osteoblasts to activate osteoclasts. The IL-1 secretion leads to further stimulation of the macrophages and amplifies the secretion of additional proinflammatory mediators maintaining a positive feedback loop. Enzymes such as collagenase can promote bone resorption by digesting the extracellular matrix, thus exposing bone mineral for osteoclastic resorption.20 All of these factors have been demonstrated in the interfacial tissue; however, there has been poor documentation of the presence of large numbers of osteoclasts at the bone–implant interface. The lack of osteoclasts is in sharp contrast to more recent studies that have carefully documented the presence of numerous macrophages containing PE particles directly in contact with the bone surface.56 Immunhistochemical studies have also confirmed the presence of enzymes within these cells that are capable of direct bone resorption. 57 Quinn and colleagues58 showed that macrophages activated by PMMA engulfment could directly resorb bone. The increased life span of the macrophage and the development of FBGCs we observed in vitro after exposure to PE particles may help to explain the increased numbers of FBGCs in contact with bone surfaces observed from retrieval specimens. Likely the chronic nature of the wear particle-induced inflammation could account for the degree of bone loss associated with aseptic loosening. There is no doubt that osteoclastic bone resorption may occur; however, evidence shows the activated macrophage is capable of resorbing the degree of bone associated with osteolysis.
Inhibition of osteoblast function and thus new bone formation is a final mechanism accelerating periprosthetic bone loss that should be considered. Goodman and associates59,60 have observed reduced new bone formation in the presence of PE particles. So, it is possible that the presence of PE particles and potential degradation products adversely influence osteoblast behaviour, decreasing new bone formation around total joint replacements.
The mechanism of periprosthetic bone loss is complex. The PE-activated macrophage is the key player; cytokines secreted by the activated macrophage may promote increased bone loss by osteoclastic bone resorption, direct bone resorption by the macrophage itself and the production of PE degradation products, which can inhibit new bone formation. Pharmacologic treatment with bisphosphonates has started in some centres with encouraging early results; however, the quality of bone formed under the influence of bisphosphonates should be carefully assessed before widespread use of these drugs can be recommended.
Future directions in osteolysis research
The future of research in periprosthetic bone loss needs to focus on 3 areas: understanding the specific sequence of cytokines produced by undifferentiated monocytes, mature macrophages and the role that T-cell cytokines play in directing PE-induced inflammation; the role of PE particle surface chemistry on macrophage activation; and characterization of PE degradation products and their effects on cells involved in bone remodelling around the implant. This information will allow the development of specific anti-inflammatory therapeutic interventions or modification of PE material processing to change the degradation-product profile preventing periprosthetic bone loss.
Acknowledgments
This work was supported by an MRC-Zimmer Industrial Grant. We acknowledge the support of the Arthritis Society and technical support from Stuart Rae and Shaomo Xing.
- Accepted February 18, 2000.
References
In this issue
{kind=link}
{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.